

The prediction of protein interaction partners (which protein interacts with which one) and the prediction of the structure of a complex between two proteins known to interact have so far been regarded as two separated issues. Physical docking programs are intended for the second goal: predict the arrangement of two protein chains of known 3D structure when they interact. These programs produce a list of complexes (docking models), representing different poses/arrangements of the two protein chains, sorted by an score representing, in the simplest case, the geometric complementarity between the two chains in these arrangements. In general, these programs have a high degree of error and the native (real) complex is usually not among the top scoring ones in this list. Additional information and constraints are usually needed to filter these lists in order to locate the native complex. This relatively poor performance, together with the intrinsically high computational requirements of these programs, precluded their usage in the search for interaction partners to date.
We have shown that, in spite of this low accuracy of the basic docking methods, in a significant number of cases they can be used to locate interaction partners within large sets of alternative structures with diverse characteristics. The distribution of raw scores of the list of complexes is shifted to better scores for the docking experiment involving a real interaction partner, than for those involving likely non-interacting chains. Paradoxically, this happens even in cases where the docking experiment involving the truly interacting chain fails in locating the real complex
A possible explanation for this observation could be related to the "energy-funnel" theory of protein interaction. According with this theory, not only the real complex between two interacting proteins has a favourable energy but there are many alternative complexes with favourable energies which help in "directing" the interaction to the real one. It is difficult to think that only the real complex has a favourable energy and that consequently the interaction process has to rely purely on random encounters until it is found, taking into account the millions of alternatives in which two proteins can face each other. Other conformations "around" the real one might also be favoured in a way that once the two chains find any of them, their way to the real complex is facilitated. That would explain the fact that, even if the real complex is not found by the docking program, these complexes are enough for shifting the energy distribution.
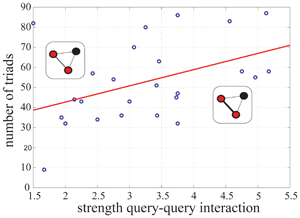
Genetic interactions are being quantitatively characterized in a comprehensive way in several model organisms. These data are then globally represented in terms of genetic networks. How are interaction strengths distributed in these networks? And what type of functional organization of the underlying genomic systems is revealed by such distribution patterns?
This work demonstrates a non-random organization of interaction strengths in genetic networks of Caenorhabditis elegans and Saccharomyces. cerevisiae, with implications for the structure of genetic buffering, the absence of strong genetic cascades, the functional role of genetic hubs and the phenotypic/evolutionary features of the genes involved. This non-random organization, common to other complex networks, could ultimately reflect how genetic variation is eventually influencing the phenotype.
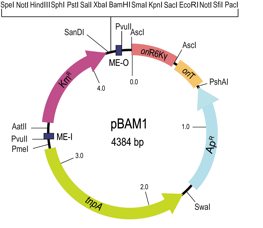
This paper reports the design and performance of two all-synthetic mini-transposon vectors, pBAM1 and pBAM1-GFP. The power of the both systemsis shown in the paper with a couple of examples.
pBAM1 plasmid map. Functional elements of the plasmid include relevant restriction sites, antibiotic markers (Ap, ampicillin, Km, kanamycin), transposase (tnpA), origin of replication (R6K), the origin of transfer region (oriT), mosaic element O (ME-O), and mosaic element I (ME-I), are shown.

In this paper, we described the function of the Crp-cAMP system of P. putida. We found that both components of the system, i.e. the genes crp and cyaA (coding for the cAMP receptor protein and adenylate cyclase, respectively) produce fully functional proteins that are not involved in the carbon catabolite repression fenomena, like the E. coli counterparts. In contrast, this system is controlling cell surface related functions, like resistance to antibiotics, toxic compounds and utilization of dipeptides as C and N source. We propose that this dissimilar functional scope of Crp-cAMP system in bacteria is a case of a molecular regulatory exaptation mechanism, i.e. the process through which a property evolved for a particular function is co-opted for a new use.
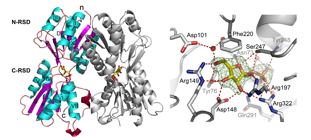
The catabolite repressor/activator (Cra) protein is a global sensor and regulator of carbon fluxes through the central metabolic pathways of Gram-negative bacteria. Our results showed that Cra regulates the PTSFru system in P. putida by binding strongly as a dimer to a single palindromic sequence within the cra/fruB intergenic region and that only F1P can lift such down-regulation.
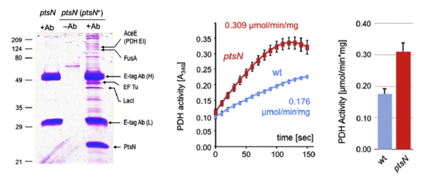
Both genetic and biochemical evidence revealed that the non-phosphorylated form of PtsN down-regulates the Pyruvate dehydrogenase activity by means of direct protein–protein interactions with AceE.This result suggests that EIIANtr takes part in the node of C metabolism that checks the flux of carbon from carbohydrates into the Krebs cycle.
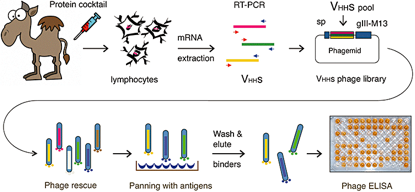
Generation of nanobody libraries workflow. The sketch summarized the main steps of the process. African camels (C. dromedarius) are inoculated with a mixture of proteins and following a 6 week period, RNA from lymphocytes is extracted, retrotrancribed to DNA and the sequences corresponding to the VHH domains amplified with suitable primers for capture in a phage display vector. This library is then separately subject to two rounds of panning on plates with the individual antigens of the original inoculation cocktail. Those M13-VHH clones that retained a strong binding activity, as detected with a phage-ELISA assay were kept for further analysis.

The A domain of the transcriptional regulator XylR of Pseudomonas putida can be evolved in the Laboratory to recreate the itinerary that goes from one exisiting effector specificity towards a new one. The figure shows amino acids that participate in such changes in effector specificity
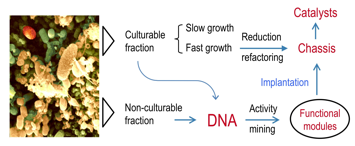
The Biotechnology of the future will increasingly depend on combining biologica activities mined from the environment (eg metagenomes) with genomic chasses engineered with the tools of Synthetic Biology.
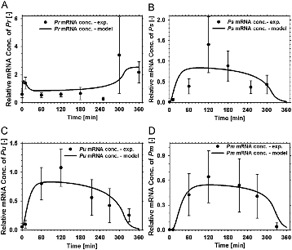
In this paper, we created a complete model of the TOL regulatory network, including both regulatory and metabolic processes, using set ordinary differential equations. Literature mining together with specifically designed experiments allows the determination of the missing kinetic parameters necessary to feed the model. Simulations of the systems allow the determination of critical regulatory steps with major contribution to the network operation. Also, predictive simulation (using in silico perturbation of the network) were performed and the results were nicely reproduced in validation experiments. We propose a general integrative strategy for the modeling of hybrid regulatory-metabolic network applicable to biodegradation process.
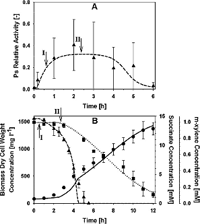
Although Boolean models are useful when few kinetic constants are available to a given network, they cannot provide high resolution information about the circuit operation. Such information is desired when regulatory/metabolic optimization is the final goal of the work, which is often the case in the field of bioengineering. In the paper, we use a model based on ordinary differential equations to try to link the dynamics of gene activation in the TOL network to biomass production and subtract consumption, final outcome of the process. The model focus only in the interplay between the two regulators of the system (XylR and XylS) and demonstrated that the outcome of the system can be successfully explained by analyze the dynamics of gene activation at this layer of the network.
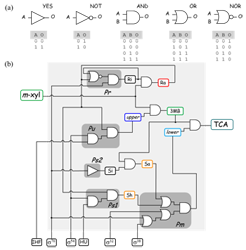
In this work we address the usage of Boolean formalism for the modeling of complex regulatory networks in bacteria. Using the pathway for degradation of toluene/m-xylene of Pseudomonas putida mt-2 as an example, we explore the potential of such formalism as a convenient tool to inspect network behavior when little kinetic information is available for the system. We argue that logic gate-driven hypothesis generation could significantly impulse the field of biodegradation.
Systems Biology Program @CNB-CSIC
c/ Darwin, 3 - 28049 Madrid - Spain
